Frederick Roth
PhD

Qualification
- Harvard Medical School, Cambridge, MA, U.S., Asst. and Assoc. Professor, 2000-2011.
- Millennium Pharmaceuticals (with Dr. Chris Sander), Cambridge, MA, U.S., Scientist, 1998-2000.
- Harvard University, Cambridge, MA, U.S., PhD in Biophysics (with Dr. George Church), 1998.
- Operon Technologies, CA, U.S., Staff Scientist, 1990-1992.
- University of California, Berkeley, CA, U.S., BA in Physics and Molecular & Cell Biology, 1990.
OTHER AFFILIATIONS
- Senior Scientist, Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto.
MY RESEARCH OVERVIEW (GO TO SCIENTIFIC OVERVIEW)
Technology to map the function of genes and variants
Members of the next human generation will routinely read their own genomes to explore one aspect of their individuality. Humanity now has the power to read the basic DNA blueprint for any species it cares to study. Unfortunately, a genome sequence is like an encyclopedia written in an alien language, and our ability to reveal DNA sequence has greatly outpaced our ability to understand it. Only a small fraction of genes are thoroughly understood, and nearly half of our genes are a complete mystery.
Progress continues to be made through the painstaking study of model organisms— cultured human cells, worms, fruit flies, mice and baker’s yeast. The success of this strategy is due to the happy accident that all of these systems share a common ancestor. New technology is needed to accelerate our study of genes, their function, how these functions interact to form living systems, and how functions and systems are altered by sequence variation.
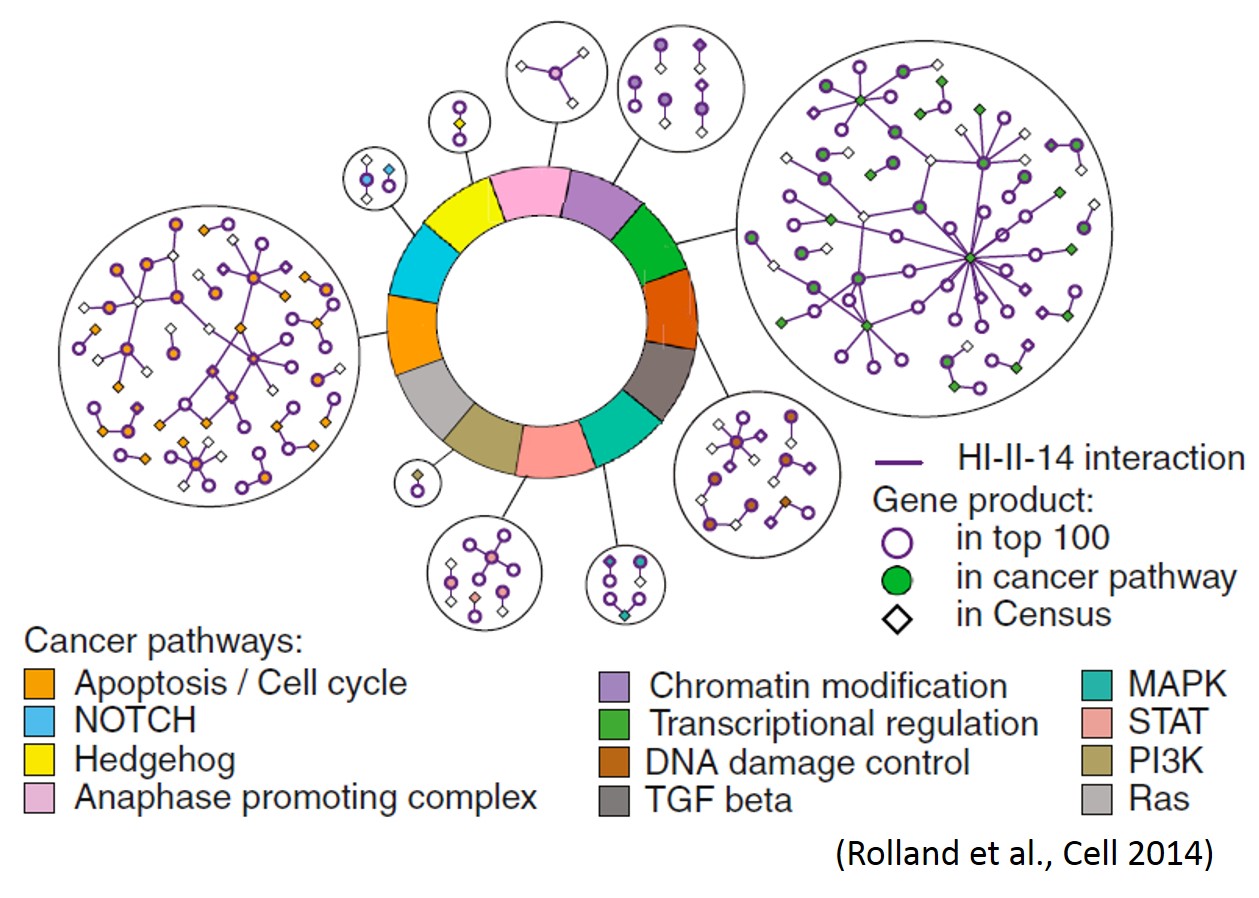
My research team is developing technology to accelerate discovery of gene functions, the pathways they encode, the relationships of genes and pathways and variation to human disease. We are exploring new ways to adapt technology for sequencing DNA to increase the scale of functional assays. For example, in a collaboration with Brenda Andrews and Charlie Boone, we are harnessing sequencing technology to systematically map the effects of multiple genetic changes in combination across multiple environments. In a collaboration with Marc Vidal, we are applying advanced sequencing technologies to efficiently measure interactions between proteins, and to identify the effects of viral proteins or cancer-causing signaling proteins on the human interaction network.
A current major challenge in medicine is interpreting the disease risk of variants in individual patient genomes. We have recently undertaken an effort to test the functional effect of essentially all possible variants in human disease genes, so that the impact of even the rarest human sequence variants can be interpreted as soon as they are first detected.
We continue to focus on developing technology to more efficiently relate genes to the functioning of living systems and human disease.
SCIENTIFIC RESEARCH OVERVIEW
Our group designs and interprets large-scale genetic and biochemical experiments to understand pathway structure and its relationship to phenotype and human disease, with a strong emphasis on protein interaction mapping, genetic interaction mapping, and identifying causal genes and variants in human disease.
A major challenge for our time is the identification of predictive and potentially actionable functional variants in individual human genomes. We are developing efficient assays to assess the functionality of human variation using complementation assays. This includes large-scale mutagenesis, selection, sequencing and analysis technologies towards building a 'lookup' table of functionality for all possible amino-acid changing mutations in human disease genes.
We are mapping protein interactions using a new “barcode fusion genetics method” based on next-generation sequencing of recombinant DNA barcodes. Current protein interaction projects include protein interaction maps amongst human centrosomal proteins, cancer-related proteins, and amongst human host targets of DNA tumourvirus proteins. We are extending these approaches to identify interactions dependent on growth condition, cell cycle and post-translational modification enzymes.
Most traits are genetically complex, so to better understand genotype/phenotype relationships we are mapping genetic interactions amongst S. cerevisiae genes under multiple growth conditions. Ongoing studies include studies of pairwise genetic interactions of DNA repair genes under multiple DNA-damaging drug conditions, engineering cell populations through which large-scale genotyping and phenotyping can uncover complex genetic interactions amongst high-order combinations of drug resistance genes. A major analysis challenge is the computational analysis of genetic interaction data to reveal redundant systems and order of action in genetic pathways. To specifically identify causal genes and alleles, we are integrating large-scale studies (including phenotype, genetic epistasis, protein-protein and transcription-regulatory interactions and sequence patterns) to quantitatively assign function to genes and guide experimentation and disease association studies.
SELECT PUBLICATIONS
- Shifting landscapes of human MTHFR missense-variant effects. Weile. et al. Am J Hum Genet. 2021 Jul 1;108(7):1283-1300.
- A reference map of the human protein interactome. Luck K et al Nature. 2020 Apr;580(7803):402-408.
- A framework for exhaustively mapping functional missense variants. Weile J et al. Mol Syst Biol. 2017 Dec 21;13(12):957