Zhaolei Zhang
PhD

Qualification
- Yale University, New Haven, CT, U.S.A., Research Fellow in Molecular Biophysics and Biochemistry, 2001-2004.
- University of California at Berkeley, CA, U.S.A, PhD in Biophysics, 2000.
- NanKai University, China, BSc in Biophysics, 1993.
MY RESEARCH OVERVIEW (GO TO SCIENTIFIC OVERVIEW)
How are genes turned on and off: the fine prints
The human genome contains over 22,000 genes, which remarkably is not too much more than the baker’s yeast or a fruit fly. So what makes humans more complex than the other organisms? The answer lies in the immensely complex and delicate ways by which the human genes are regulated, or turned on and off. Simple glitches in these regulations will result in a protein being made at an abnormal level or at wrong time or wrong place, which in turn can cause diseases such as cancer or neurological disorders. In our laboratory, we are particularly interested in studying two types of gene regulations: those mediated by microRNAs and by DNA methylation. We use computers to compare the molecular information gathered from healthy people and people with diseases, aiming to find what glitches in the genome cause these diseases. Our ultimate goal is to use the information we gained to design better disease diagnostics tools and more precise medical treatments.
Visit Dr. Zhaolei Zhang's Discover Research profile to learn more.
SCIENTIFIC RESEARCH OVERVIEW
My lab is a computational biology lab. We develop novel and efficient computational software to analyze genomic data to understand how human genes are regulated and how mutations in proteins and in regulatory elements can cause diseases. We are particularly interested in developing computational tools that have direct impact on biomedical diagnostics. We work closely with experimental and clinical colleagues on these projects.
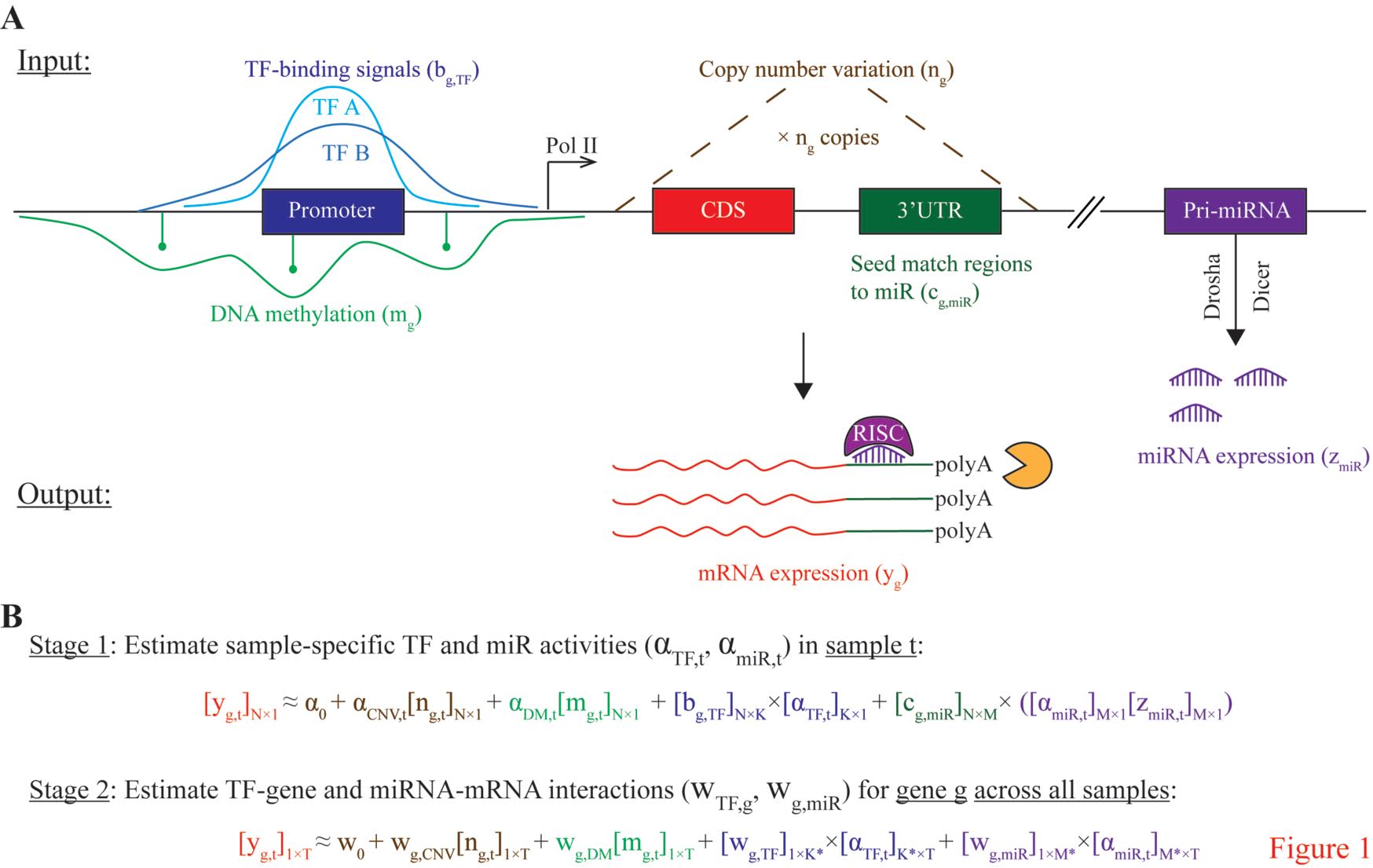
We have developed novel microRNA target prediction methods that integrate sequence complementarity with gene expression data. We applied these methods in the analysis of pan-cancer datasets and identified microRNA-gene pairs that are important in individual types of cancer. We have also developed a linear regression framework that can model the effect of transcription factors, DNA methylation, CNVs and microRNAs on gene expression. We are also studying how microRNA targeting can be weakened or enhanced by mRNA methylation.
Together with clinical researchers, we develop statistical methods to analyze population based DNA methylation datasets to identify epigenetic changes that are associated with cancer or neurological diseases such as depression or autism. Particularly we are exploring whether such epigenomic changes can be used as early diagnostics tools.
We are also developing software tools that can efficiently analyze exome sequences and make accurate variant calls and variant annotations. We then apply the software SNPdryad to predict whether a coding variant (SNP) is likely to be deleterious.
SELECT PUBLICATIONS
- Potential microRNA-mediated oncogenic intercellular communication revealed by pan-cancer analysis. Li Y. & Zhang Z. Scientific Reports. 2014 Nov 18;4:7097.
- Regression analysis of combined gene expression regulation in acute myeloid leukemia. Li Y, Liang M, Zhang Z. PLOS Computational Biology, 2014 Oct 23.
- Identifying mRNA Sequence Elements for Target Recognition by Human Argonaute Proteins. Li J, Kim T, Nutiu R, Ray D, Hughes TR and Zhang Z. Genome Research 2014 May;24(5):775-85.